ClassyFlu: Classification of Influenza A Viruses with Discriminatively Trained Profile-HMMs – PLOS ONE
See on Scoop.it – Virology and Bioinformatics from Virology.ca
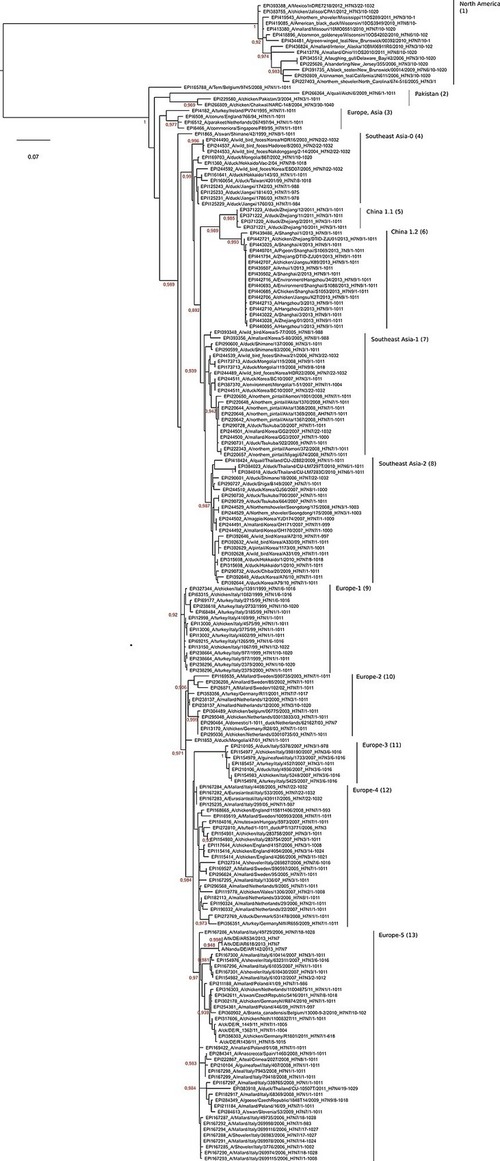
Accurate and rapid characterization of influenza A virus (IAV) hemagglutinin (HA) and neuraminidase (NA) sequences with respect to subtype and clade is at the basis of extended diagnostic services and implicit to molecular epidemiologic studies. ClassyFlu is a new tool and web service for the classification of IAV sequences of the HA and NA gene into subtypes and phylogenetic clades using discriminatively trained profile hidden Markov models (HMMs), one for each subtype or clade. ClassyFlu merely requires as input unaligned, full-length or partial HA or NA DNA sequences. It enables rapid and highly accurate assignment of HA sequences to subtypes H1–H17 but particularly focusses on the finer grained assignment of sequences of highly pathogenic avian influenza viruses of subtype H5N1 according to the cladistics proposed by the H5N1 Evolution Working Group. NA sequences are classified into subtypes N1–N10. ClassyFlu was compared to semiautomatic classification approaches using BLAST and phylogenetics and additionally for H5 sequences to the new “Highly Pathogenic H5N1 Clade Classification Tool” (IRD-CT) proposed by the Influenza Research Database. Our results show that both web tools (ClassyFlu and IRD-CT), although based on different methods, are nearly equivalent in performance and both are more accurate and faster than semiautomatic classification. A retraining of ClassyFlu to altered cladistics as well as an extension of ClassyFlu to other IAV genome segments or fragments thereof is undemanding. This is exemplified by unambiguous assignment to a distinct cluster within subtype H7 of sequences of H7N9 viruses which emerged in China early in 2013 and caused more than 130 human infections. http://bioinf.uni-greifswald.de/ClassyFlu is a free web service. For local execution, the ClassyFlu source code in PERL is freely available.
See on www.plosone.org