The Gene Results Table
The Gene Results Table Main Window
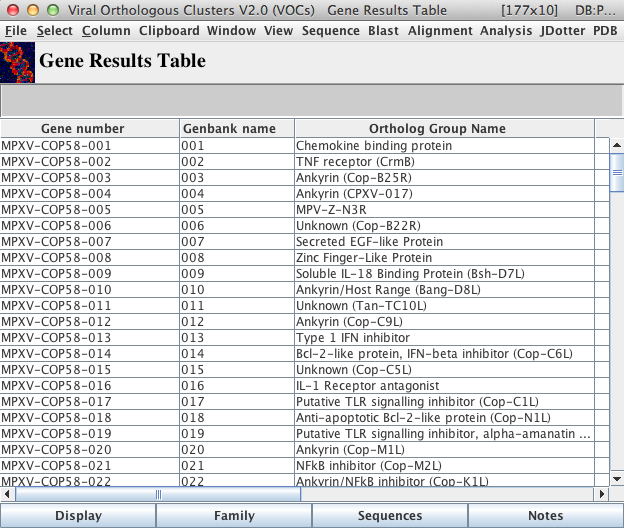
The “Display” button found at the bottom of the table will display more in-depth detail of all genes selected within the main window of the table.
The “Family” button found at the bottom of the table will display all genes found within the same ortholog groups as the genes selected in the main window of the table.
The “Sequences” button will display both the gene sequence and corresponding protein sequence for all selected genes.
The “Notes” button will display one window containing any curated notes associated with the selected genes.
The Gene Results Table Menu Bar
Gene and protein analysis tools like Clustalw, BLAST, JDotter etc. may be accessed using the menu bar of this table. All programs will run using the gene sequences selected in the table.
![]()
The “View” Menu
View InterPro and GO references, PDB structure assignments, annotated notes and gene details for all genes you have selected.
The “Sequence” Menu
“Search“
Conduct a regular expression sequence search or a fuzzy sequence search. For a list of IUPAC allowable one-letter amino acid/nucleotide codes, visit the “Amino Acid Constraints“/”DNA Constraints” sections of The Sequence Filter page.
For the regular expression sequence search, enter the desired string of amino acids/nucleotides in the search field to have VOCs search the selected gene sequences for exact matches. Remember this type of search search will only return hits that match the exact value of the one-letter amino acid/nucleotide code rather than the values to which the code translates (ie. for the amino acid code “J”, VOCs will return matches containing the actual letter “J” but NOT “I” or “L” (to which “J” is equivalent)).
If you wish to allow some margin of error in a sequence search for your selected genes, you may choose to conduct a fuzzy sequence search where you may select the number of mismatches you are willing to allow. For this type of search, entering the one-letter nucleotide code will return hits that not only match the one-letter code but also match the values to which it translates (ie. for the nucleotide code “R”, VOCs will return matches containing “A” or “G” (to which “R” is equivalent)). The analogous search function for amino acids in the fuzzy search is not yet available but will be available in the near future.
- “DNA Sequence(s)” will search every nucleotide between the first gene and the last gene you select
- “Upstream Sequence(s)” will search 100 bp upstream of the start codon and the first 10 nucleotides of the coding regions (ATG inclusive) you select regardless of any coding sequence that may be found within the 100 bp found upstream of the start codon.
- “Protein Sequence(s)” will search all the amino acids of the corresponding proteins for the selected genes
- “Genomic DNA Sequence(s)” will only search every nucleotide of the coding regions of each gene you select
“View“
In one window, this will display the either the DNA sequences, Upstream DNA sequences, corresponding Protein sequences or Genomic DNA sequences for all genes you have selected in the Gene Results Table main window.
“Save“
This will save the data you are able to see using the “View” function above.
The “BLAST” Menu
Should you wish to use BLAST for a gene or a protein, you may do so here. You may choose to use tBLASTn, BLASTx, BLASTp, family BLASTp, BLASTn or psiBLAST to run your query of interest against our own database or NCBI’s database. There are several views available from which to choose: text, HTML, mview and table view.
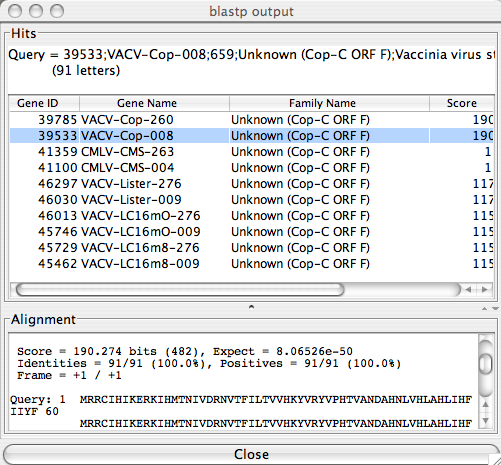
The BLAST Table View
The BLAST table view lists all hits reported from a BLAST run in a table format. The alignment details can be viewed by clicking on a row in the summary table. Any column in the table can be sorted by simply clicking on the column header.
The Family BLASTp
Ortholog groups (gene families) can be defined as groups of genes that share significant sequence similarity. In VOCs, ortholog group assignment is partially based on the E value obtained by BLASTp. This method clusters proteins that are very similar to each other (for example, genes of different strains of the same virus), but often does not co-cluster more distantly related proteins. “Run family BLAST against VOCs DB” creates a BLASTp output of the genes you select against the genes within the ortholog groups of the database you are working in. All results are summed up in a tabular format. The assumption is that if all or most proteins of genes in one ortholog group are similar to all or most proteins of genes in another ortholog group, those two ortholog groups are said to be significantly similar. The similarity is assessed using BLAST scores and E-values.
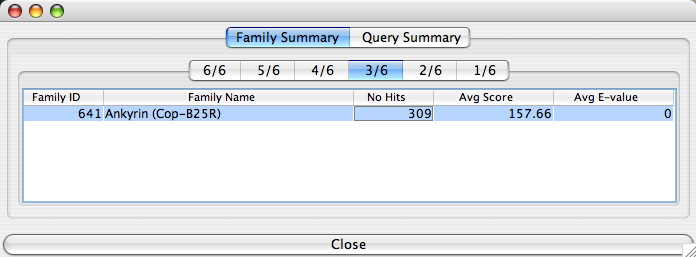
The “Family Summary” tab of this table contains multiple tabs labelled “n/N” where “N” is the number of genes you selected for the family BLASTp and “n” is a number from 1 to “N” inclusive. This number (“n”) represents the number of proteins (corresponding to your selected genes) that produced positive matches to proteins of genes in other ortholog groups according to your BLASTp query settings. For instance, if you select VACV-LC16m8 (genes 005, 006, 007, 008, 009 and 010) for the family BLASTp query, the table above will be displayed. The tab “3/6” means that 3 of the 6 proteins matched with proteins of genes in the ortholog groups listed in the main window; in this case there is only one ortholog group listed (“Ankyrin(Cop-B25R)”). “# Hits” indicates the cumulative number of hits BLASTp returned for all 3 proteins. “Avg Score” is the average BLAST score calculated using the scores for the hits of all 3 proteins. “Avg E-value” is the average BLAST E-value calculated using E-values corresponding to the hits returned for all 3 proteins. For a more detailed view of this information, click on the line corresponding to the ortholog group of interest. Once you do, you will get something similar to the following table:
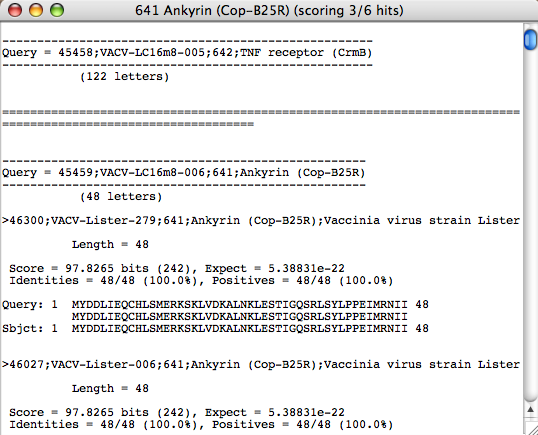
This view of the BLASTp alignments will display all of the proteins of the genes you selected for the BLASTp query and any corresponding hits. Under each gene name is the length of the protein in brackets. In this case, the first gene is VACV-LC16m8-005. Directly under this name is “(122 letters)” which means the protein of this gene consists of 122 amino acids. All BLAST alignment information and values are displayed in the standard BLAST output format.
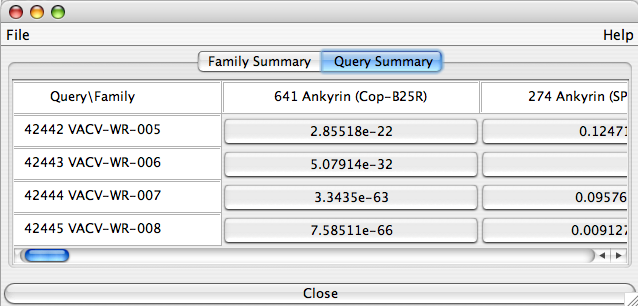
The “Query Summary” tab displays all ortholog groups (accross the first row) that reported at least one BLASTp hit on any protein of your selected genes (listed down the first column). The E-values for the BEST hit in the corresponding ortholog group are displayed in cells. All alignments specific to one ortholog group can be viewed by clicking on the appropriate cell.
The “Alignment” Menu
Here you can create alignments of DNA and protein sequences using programs like Clustal Ω , T-Coffee, Needle, JAligner, MUSCLE (only applies to protein sequences) and Near-optimal Align. The alignment output will differ depending on which alignment program is selected, however, all alignments will be displayed in a Base-By-Base window. If the option “unaligned” is selected, the sequences will be exported to Base-By-Base without being aligned.
The “Analysis” Menu
You can create a hydrophobicity plot with Hydrophobicity Grapher for multiple genes; all plots will be displayed on the same graph and all the amino acid sequences will be displayed in a window immediately below. The hydrophobicity value found on the Y-axis of the graph depends on the scale selected from the drop-down menu above the main window. The hydrophobicity scales available from which to choose are: Kyte-Doolittle (default), Hopp-Woods and Parker-Guo-Hodges.
If you would like to view nucleotide composition in a graphical format, select “DNA Grapher“, this will create a DNA skew graph with GraphDNA.
The “JDotter” Menu
Use JDotter to compare the similarity between multiple genes using one dotplot graph. From this menu, you may choose to create a dotplot to map the similarity of the selected genes’ corresponding protein sequences, DNA sequences (coding region) or upstream sequences.