j-CODEHOP How-to Guide
Variables in j-CODEHOP
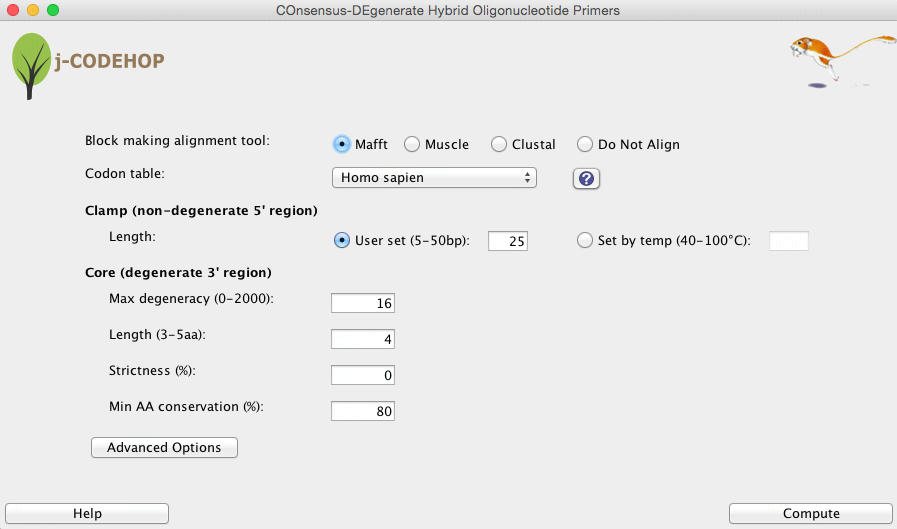
Alignment tools
Muscle: Muscle is recommended for multiple sequence alignments of amino acid sequences; it performs global alignments with relatively good accuracy and speed [1].
ClustalO: ClustalO has faster performance on very large sets of sequences [2].
Codon Table
Codon tables form several organisms can be selected from in this pull-down menu. This allows the user to apply species-specific codon usage bias on how the amino acid position-specific scoring matrix (PSSM) will be back-translated to DNA. Codon usage tables for several organisms can be selected or Custom codon usage tables can be created. Select the “?” button to learn more about Custom codon tables and codon table sources.
Clamp
Length: Clamp length can either be selected by user, or determined by annealing temperature. A higher annealing temperature results in a longer clamp length. The clamp length may be adjusted without increasing degeneracy.
Core
Max degeneracy: measures how many different sequences are specified by the primer. The degeneracy of each position is the number of nucleotides appearing in it (1 to 4). The degeneracy of the primer is the product of the degeneracies of each position. A smaller degeneracy increases the clamp score, which indicates the quality of the match between the 5′ non-degenerate clamp and the sequence block given a codon usage table. However, if no primer is identified, the max degeneracy may be increased, which may identify highly conserved motifs that contain amino acids with higher codon degeneracies.
Length: Usually 3-5 amino acids long, from which all of the possible nucleotide sequences are used for primers. In general, the longer the core length, the fewer CODEHOPs produced since more amino acids must match up.
Strictness: Is value between 0 and 1 specifying a level of occurrence for nucleotide inclusion. Strictness of 0 means that all nucleotides that actually appear in the position are included in the primer. By increasing the degeneracy ‘strictness’, lower abundance nucleotides can be excluded from the 3’ degenerate core.
Min AA conservation: Changes the minimum amino acid conservation in the primer core; a smaller amino acid conservation in the core will in general result in an increase in primers.
Advanced Options:
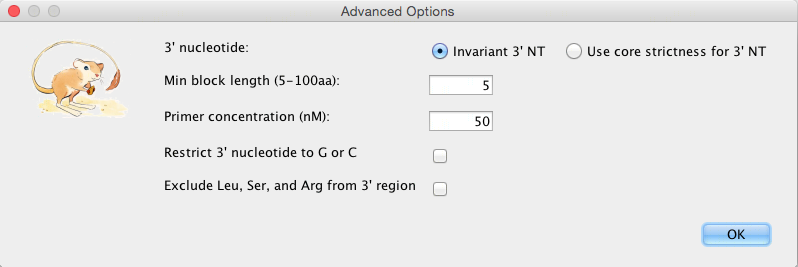
3′ nucleotide: “Invariant 3′ NT” disregards the strictness option and will use a strictness of 0 (only a nucleotide that appears in the primer), while “Use core strictness for 3′ NT” will use the inputted strictness for choosing the 3′ primer. A 3’ NT with a strictness of 0 makes the primer more stable.
Min block length: A block is an ungapped multiple alignment segment, the minimum length of a block within which a group of primers will be generated can be adjusted.
Restrict 3’ nucleotide to G or C: G-C bonds have three hydrogen bonds, making primers with a 3’ G or C more stable, and therefore function better.
Exclude Leu, Ser, and Arg from 3’ region: These amino acids have the highest degeneracy (each being 6). Excluding them excludes primers with a high degeneracy due to these amino acids.
References:
[1] Edgar R C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acid Res. 32(5): 1792-1797.
[2] Sievers F, and Higgins D G. (2013) Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences, Methods in Molecular Biology. 1079: 105-116.