Base-By-Base Documentation
Getting to know Base-by-Base
A brief introduction to the functions of Base-By-Base and its main windows.
Main Window
The components of the main window.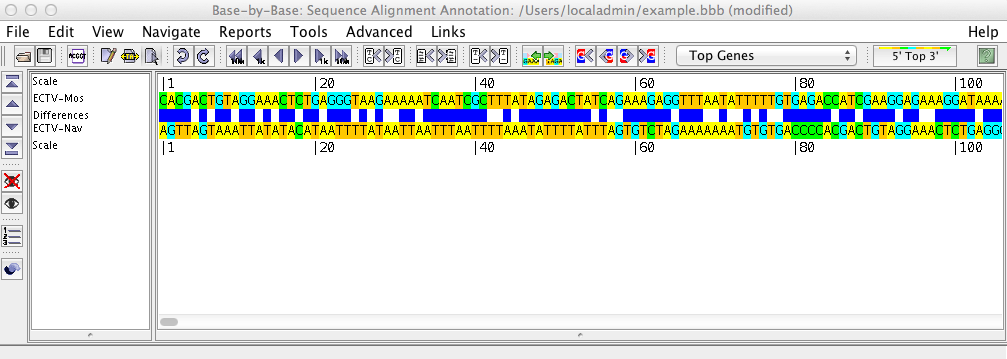
The main window consists of four sections:
The Menu bar
![]()
The items in the menu bar can be used for loading sequences into Base-By-Base, manipulating and aligning the loaded sequences, viewing reports about the sequences, and opening related websites.
The Tool bar
The tool bar contains buttons for performing the most common actions.![]()
At the left-most edge of the toolbar are two vertical lines which can be used to drag the toolbar to a different location in the Base-By-Base window or outside of the window entirely. To the immediate right of all the main toolbar buttons is a drop-down list of genes found in the sequences (that reads “Top Genes” by default). To jump ahead to a gene of interest, select it from this menu. It is also possible to switch between the top strand and the bottom strand views by clicking on the next box (that reads ” 5′ Top 3′ ” by default). To the right of this box is a square button with a question mark that links to the help pages for Base-By-Base (this manual).
In order from left to right, the toolbar buttons and corresponding functions are:![]() Load a previously-saved alignment or individual sequences to be aligned into Base-By-Base.
Load a previously-saved alignment or individual sequences to be aligned into Base-By-Base.![]() Save the current alignment to a file.
Save the current alignment to a file.![]() Adjust the display area.
Adjust the display area.![]() Set mouse mode to Edit. You can insert, resize or remove gaps by using this mode. To do so, click on the residue you would like to be at the end of the gap and drag your mouse horizontally until the desired gap length is attained.
Set mouse mode to Edit. You can insert, resize or remove gaps by using this mode. To do so, click on the residue you would like to be at the end of the gap and drag your mouse horizontally until the desired gap length is attained.![]() Set mouse mode to Block Glue. You can shrink or remove gaps by using this mode. To do so, click on the residue immediately following the gap and drag your mouse to the left until the gap is the desired length.
Set mouse mode to Block Glue. You can shrink or remove gaps by using this mode. To do so, click on the residue immediately following the gap and drag your mouse to the left until the gap is the desired length.![]() Set mouse mode to Select. This feature will allow you to highlight/select a region to which you would like future parameters to apply. It is not possible to insert, resize or remove gaps with this feature.
Set mouse mode to Select. This feature will allow you to highlight/select a region to which you would like future parameters to apply. It is not possible to insert, resize or remove gaps with this feature.![]() Undo the last change to a sequence.
Undo the last change to a sequence.![]() Redo the last change to a sequence.
Redo the last change to a sequence.![]() Jump backwards (left) 10kbp.
Jump backwards (left) 10kbp.![]() Jump backwards (left) 1kbp.
Jump backwards (left) 1kbp.![]() Jump backwards (left) by 20 units.
Jump backwards (left) by 20 units.![]() Jump forwards (right) by 20 units.
Jump forwards (right) by 20 units.![]() Jump forwards (right) 1kbp.
Jump forwards (right) 1kbp.![]() Jump forwards (right) 10kbp.
Jump forwards (right) 10kbp.![]() Go to the last difference between the sequences. If all but one sequence contains an identical residue at the same position, it is still considered a difference.
Go to the last difference between the sequences. If all but one sequence contains an identical residue at the same position, it is still considered a difference.![]() Go to next difference between the sequences. If all but one sequence contains an identical residue at the same position, it is still considered a difference.
Go to next difference between the sequences. If all but one sequence contains an identical residue at the same position, it is still considered a difference.![]() Go to the previous comment found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the comment corresponds will be ligned up with the left-hand edge of the sequence window.
Go to the previous comment found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the comment corresponds will be ligned up with the left-hand edge of the sequence window.![]() Go to the next comment found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the comment corresponds will be ligned up with the left-hand edge of the sequence window.
Go to the next comment found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the comment corresponds will be ligned up with the left-hand edge of the sequence window.![]() Go to the previous sequence gap found in any of the sequences visible in the sequence window.
Go to the previous sequence gap found in any of the sequences visible in the sequence window.![]() Go to the next sequence gap found in any of the sequences visible in the sequence window.
Go to the next sequence gap found in any of the sequences visible in the sequence window.![]() Go to the previous primer found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the primer corresponds will be ligned up with the left-hand edge of the sequence window.
Go to the previous primer found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the primer corresponds will be ligned up with the left-hand edge of the sequence window.![]() Go to the next primer found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the primer corresponds will be lined up with the left-hand edge of the sequence window.
Go to the next primer found for any of the sequences visible in the sequence window. The first residue of the sequence fragment to which the primer corresponds will be lined up with the left-hand edge of the sequence window.![]() Go to the previous gene found in any of the sequences visible in the sequence window. The first residue of the gene will be lined up with the left-hand edge of the sequence window.
Go to the previous gene found in any of the sequences visible in the sequence window. The first residue of the gene will be lined up with the left-hand edge of the sequence window.![]() Go to start of gene.
Go to start of gene.![]()
Go to end of gene.![]() Go to the next gene found in any of the sequences visible in the sequence window. The first residue of the gene will be lined up with the left-hand edge of the sequence window.
Go to the next gene found in any of the sequences visible in the sequence window. The first residue of the gene will be lined up with the left-hand edge of the sequence window.![]()
Drop down menu to select a specific gene.![]() View other strand.
View other strand.
The Sequence Toolbar
The sequence toolbar buttons will allow you to alter the view of the list of sequences in the sequence window.
In order from top to bottom, these buttons perform the following actions:![]() Move the marked sequence(s) to the top of the list.
Move the marked sequence(s) to the top of the list.![]() Move the marked sequence(s) up one position in the list.
Move the marked sequence(s) up one position in the list.![]() Move the marked sequence(s) down one position in the list.
Move the marked sequence(s) down one position in the list.![]() Move the marked sequence(s) to the bottom of the list.
Move the marked sequence(s) to the bottom of the list.![]() Hide the marked sequence(s) from view.
Hide the marked sequence(s) from view.![]() Open a window that will allow you to select the sequences you wish to hide from a list of all sequences in the window. This option is particularly useful when you are working with a large number of sequences within Base-By-Base.
Open a window that will allow you to select the sequences you wish to hide from a list of all sequences in the window. This option is particularly useful when you are working with a large number of sequences within Base-By-Base.![]() Toggle on/off the 3 frame amino acid translation for the marked sequences.
Toggle on/off the 3 frame amino acid translation for the marked sequences.
The Sequence Window
The sequence shows the loaded sequences, base by base, in an editable format. Using the menu and the toolbar buttons, you can perform various operations on the sequence such as inserting or removing gaps, adding primers, genes, or comments, or aligning the sequences with Muscle, ClustalΩ, or Mafft.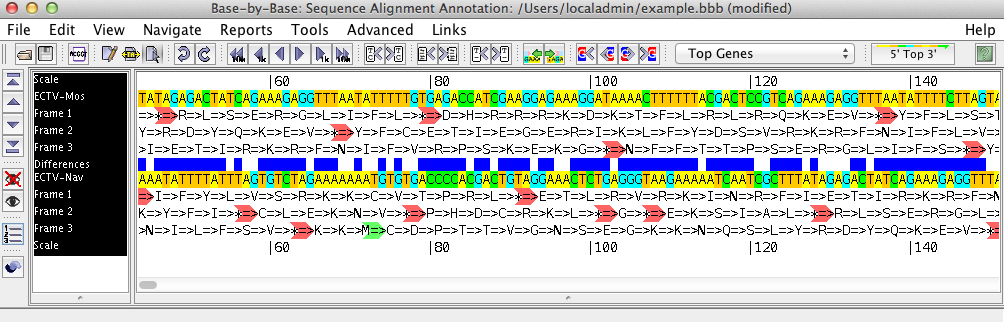
The left side of the sequence list shows the names of the displayed sequences, and gives headings for the various rows displayed above/beneath them (such as the scale, amino acid translation frames, primers, comments, etc.) Clicking on one or more sequence names will select these sequences; all changes then made will affect these sequences only.
The right side of the sequence list contains the nucleotide sequence. By clicking on one of the three mouse mode buttons in the toolbar (Edit, Block Glue, and Select) you can insert/remove gaps, glue alignments together, and select regions of the sequence on which to perform other operations.
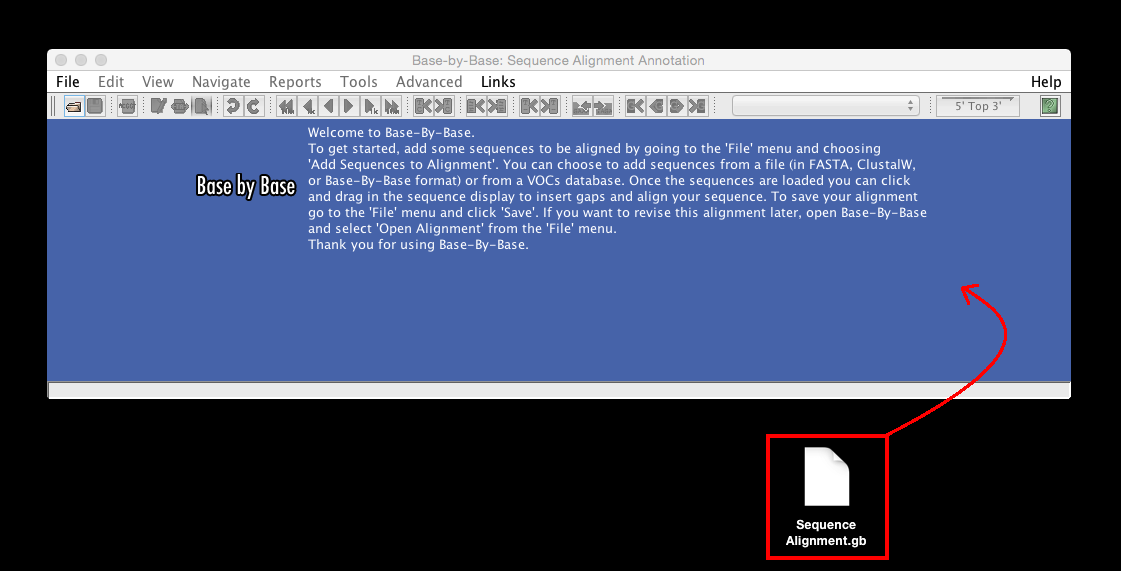
“Drag to add sequence to alignment or open alignment” to load a previously-saved alignment or individual sequences to be aligned, simple drag in the file/files onto the initial Base-By-Base sequence window (The local file formats that are accepted are : FASTA format, Base-By-Base format, Clustal format (.aln), Genbank format, and EMBL format).
The Menu Bar
A description of all features listed within the menu bar of Base-By-Base.![]()
The items in the menu bar can be used for loading sequences into Base-By-Base, manipulating and aligning the loaded sequences, viewing reports about the sequences, and opening related websites.
The File Menu
“New Alignment” will open a second Base-by-Base window to allow you to work with more than one multiple sequence alignment at the same time.
“Add Sequences to Alignment” will allow you to add the sequences you wish to align to the Base-By-Base sequence window. Sequences can be imported from the VOCs database or from a local file (the local file formats that are accepted are: FASTA format, Base-By-Base format, Clustal format (.aln), Genbank format and EMBL format). Sequences can also be imported by dragging a sequence file from your computer into the BBB window.
“Open Alignment” loads previously aligned sequences into Base-By-Base (the file formats that are accepted are: FASTA, Base-By-Base, Clustal-formatted, Genbank and EMBL).
“Remove Sequences” removes any selected/highlighted sequences from the sequence window.
“Save, Save As…” will save the sequence alignment.
“Save Primers…” will save the primers of the sequence that is selected.
“Export Selection (Marked Sequences)…” exports the marked sequences to a file in FASTA, BBB, ClustalW or Primer format.
“Export Selection (All Sequences)…” exports all sequences to a file in FASTA, BBB, ClustalW or Primer format.
“Export Alignment Image” exports the image to a file in jpeg or png format. In the window that pops up, image settings such as the start and stop position to be included in the image, image width, spacing and scaling can all be modified prior to saving the image.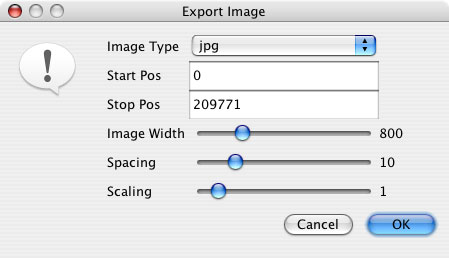
“Delete Gap Columns and Export…” will export all selected sequences to one FASTA or ClustalW file. Gaps are removed prior to exporting to the file.
“Preferences” pulls up an “External Application Preferences” window (if “Display Preferences” is selected) in which the parameters for BLAST, ClustalΩ, and Muscle can be set for use in Base-By-Base. When “Display/Usability Preferences” is selected, an “Edit Preferences” window pops up – within which you can set a number of parameters for the functionality of Base-By-Base itself.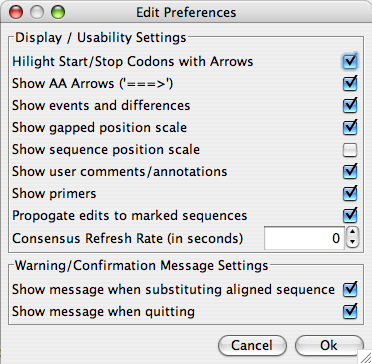
The following is a description of each of the preference options:
| Preference | Action |
|---|---|
| Highlight Start/Stop Codons with Arrows | If checked, the start and stop codons are highlighted in green and red (respectively) in the 3 frame sequence translation. |
| Show AA Arrows | If checked, an arrow “=>” will follow each one-letter aa code to better visualize the end of it’s corresponding codon. |
| Show events and differences | If checked, a row of colored region representing the differences between residues is displayed for each sequence. |
| Show gapped position scale | If checked, a fixed scale becomes visible at the top of the sequence window. This is particularly useful when gaps are present. |
| Show sequence position scale | If checked, then a dynamic scale is visible for each sequence. This is particularly useful when gaps are present. |
| Show user comments/annotations | If checked, user comments are displayed. |
| Show Primers | If checked, primers are displayed. |
| Propogate edits to marked sequences | If checked, when an edit operation is performed on one sequence, it will also be performed on all other marked sequences. |
| Consensus Refresh Rate (in seconds) | When a consensus is displayed, this setting is used to choose the length of the delay between updates of the consensus. If this is set to 0, the consensus is only refreshed when the user chooses to refresh it. |
| Show message when substituting aligned sequence | After an alignment is made using Muscle, ClustalΩ, or Mafft, a new window is displayed with the aligned sequence. If this is checked, a warning message will be displayed when you choose to accept the alignment. |
| Show message when quitting | If this is checked, when you choose to close Base-By-Base, you will be prompted to confirm that you would like to quit. |
“Close Alignment” will remove all sequences from the sequence window without saving any part of the alignment.
“Quit” exits Base-By-Base.
The Edit Menu
“Undo/Redo” will undo/redo the last action.
“Copy” will copy the highlighted region(s).
“Mark All Sequences“/”Unmark Sequences” will mark/unmark all sequences.
“Select Whole Sequence” selects the entire sequence for all marked sequences.
“Select Region” brings up a window within which you can define the parameters for the region(s) you would like to select. The sequence(s) of interest can be selected from the drop-down menu.
“Insert Gaps” inputs a specified number of gaps at the region selected in the sequence window. In order for this feature to be functional, the sequence of interest must be marked.
“Remove Gaps in Selected Region(s)” eliminates all gaps within the region that is selected in the sequence window.
“Remove Selected Sequence Region(s)” deletes all nucleotides/amino acids within the region selected in the sequence window. In order for this feature to be functional, the sequence of interest must be marked.
“Remove Identical Sequence(s)” deletes any duplicate sequences in the alignment. All but one sequence from sequence sets that are exactly identical will be deleted. All matching nucleotides/amino acids as well as any gaps must be located at identical positions in all sequences in order for sequences to be considered identical.
“Remove Not Selected Sequence Region(s)” deletes all nucleotides/amino acids within the region that is not selected in the sequence window. In order for this feature to be functional, the sequence of interest must be marked.
“Remove Gapped Columns in Selected Region” will delete any gaps that are in identical positions of all sequences in the sequence window. Regions do not have to be selected in the sequence window and sequences do not need to be marked.
The View Menu
“Comparison Method” gives you the option to highlight the sequences in varying ways: either “pairwise comparison” (an alignment of each sequence with the one directly below it), “against consensus” (an alignment of each sequence against the calculated consensus for all sequences listed in the sequence window) or “against top sequence” (an alignment of each sequence against the top sequence of the sequence window).
“Set Display Area…” will display a pop up window within which you can define the visible portion of the sequences with which you would like to display/work.
“Show/Hide Sequences…” either displays or hides certain sequences in the sequence window to allow you to work with a subset of the sequences initially added to the Base-By-Base alignment.
“Color Scheme” allows you to change the color of the nucleotides/amino acids based on differences between: nucleotides/amino acids, similarity, hydrophobicity, BLOSUM62 score, PAM250 Score, Percent ID or Custom. For amino acid sequences, a custom color scheme search will allow you to selectively choose an amino acid, and the function will color in the sequences if present. Normally, it does not function for a DNA sequence file, but is a useful trick if “N’s” are present in the sequence as Base-By-Base will believe the file to be a protein file and “Ns” can be highlighted.
“Difference Colors” allows you to change the colors used to show differences:
“Insertions/Substitutions/Deletions” shows insertions in green, deletions in red and substitutions in blue (the default)
“Nucleotide Differences” shows substitutions by the color of the substituted nucleotide; yellow for A, green for C, cyan for G and orange for T. Blue is used where the substitution is not apparent (for instance in consensus comparisons where there is no consensus, or substitutions to X or U). This mode only works for Nucleotide sequences at this time.
“Display Consensus” will allow you to show or hide the consensus alignment/sequence in a window directly below the sequence window.
“Refresh Consensus” will refresh the consensus alignment/sequence after changes have been made to the sequences or alignment in the sequence window above.
The Navigate Menu
“Go To Location…” pulls up a window that gives you the ability to jump to a desired position in the sequence you select from the drop-down menu in the window. The position value can be based on the absolute value or the gapped position depending on what you select.
The rest of this menu is comprised of buttons found in the toolbar. For the function of these options, please see the Toolbar section.
The Reports Menu
“Neighbor Joining Tree” will create a tree graph comparing the relationship between sequences in the sequence window based on the number of expected substitutions per site. The tree will be displayed in a new window.
“Clustering Tree” will create a tree graph comparing the relationship between sequences in the sequence window. The types of clustering trees available are: complete linkage, single linkage, UPGMA or WPGMA. The tree graph will be displayed in a new window.
“View CDS Statistics” will display a “CDS Event Statistics” window for 2 marked sequences of your choice. In the window, you will find statistical information about each gene and the differences between it and its corresponding gene in the 2nd sequence. The statistical fields listed in the table are as follows: gene name, gene location (strand), ORF start, ORF stop, length, aligned length, differences, % differences, substitutions, insertions, deletions, changes to the region located 200bp upstream differences, amino acid changes, silent changes, corresponding gene (counterpart), counterpart length (length) and length difference. The data in the columns corresponding to substitutions, deletions or insertions is interpreted as follows: the first number represents the total number of substitutions/insertions/deletions. The first numEach consecutive set of numbers represents the “type” of substitution/deletion/insertion repeat (number of consecutive substitutions/insertions/deletions within the sequence); immediately followed by a number in brackets corresponding to the total number of times this type of substitution/insertion/deletion occurs in the selected region for which the CDS Statistics were generated. Each column can be sorted by clicking on the respective header. This table can be exported to an HTML or CSV file.
“View Multi Genome Comparison Statistics” (also known as MGC statistics) will generate a table with a list of statistics for multiple genome alignments. Each column can be sorted by clicking on it’s corresponding header. The statistical fields are as follows: reference sequence location (the number represents the position of a nucleotide change between the “master” sequence and the other sequence(s) – any gaps will be included in the calculation of this position), nucleic acid (na) substitution (the value of the nucleotide substitution whose position is listed in the previous column – a result will not be displayed if any of the sequences contain a gap for this position), feature affected (this lists the name(s) of the gene(s) that is affected by the substitution), reference aa location (this will list the amino acid in the reference gene, the amino acid in the affected gene as well as both of the genes direction/strand location), DNA sequence (this will show the aa change followed by the change in the codon nucleotide(s); silent substitutions will be marked with an “x”).
“View Primer Report” will display a “Primer Report” window for the marked sequences of your choice. In the window, you can find statistical information for all primers corresponding to these sequences depending on the information contained in the file from which you uploaded the primers or which information you entered when you added a primer. The statistical information that will be displayed for the primers is as follows: sequence, name, comment, location, melting temperature, start, stop, length, strand and genome name (species).
“View Event Breakdown” calculates the “events” such as percent difference between the sequences in the sequence window and displays the breakdown of these “events” in a pop-up window. These “events” include: the number and type of substitutions and/or amino acid changes and number of deletions.
“View Gap Count” will display the number of gaps that exist in all loaded sequences independent of whether or not they are visible in the sequence window.
“Pairwise Alignment” will export the current alignment of two marked sequences to a separate window. A new selection of two marked sequences will export the results to a new window. Matching residues will be connected with vertical lines. This is particularly useful if you are working with a large number of sequences where it is difficult to view the alignment of two specific sequences of the group. It is also useful to use this feature to quickly compare, side-by-side, the difference between sequences with and without an alignment program and/or using varied alignment programs.
“Consensus Report” will display a pop-up window with the consensus sequence for all sequences visible in the sequence window.
“Sequence similarity graph” will display a pop-up window with a similarity graph between sequences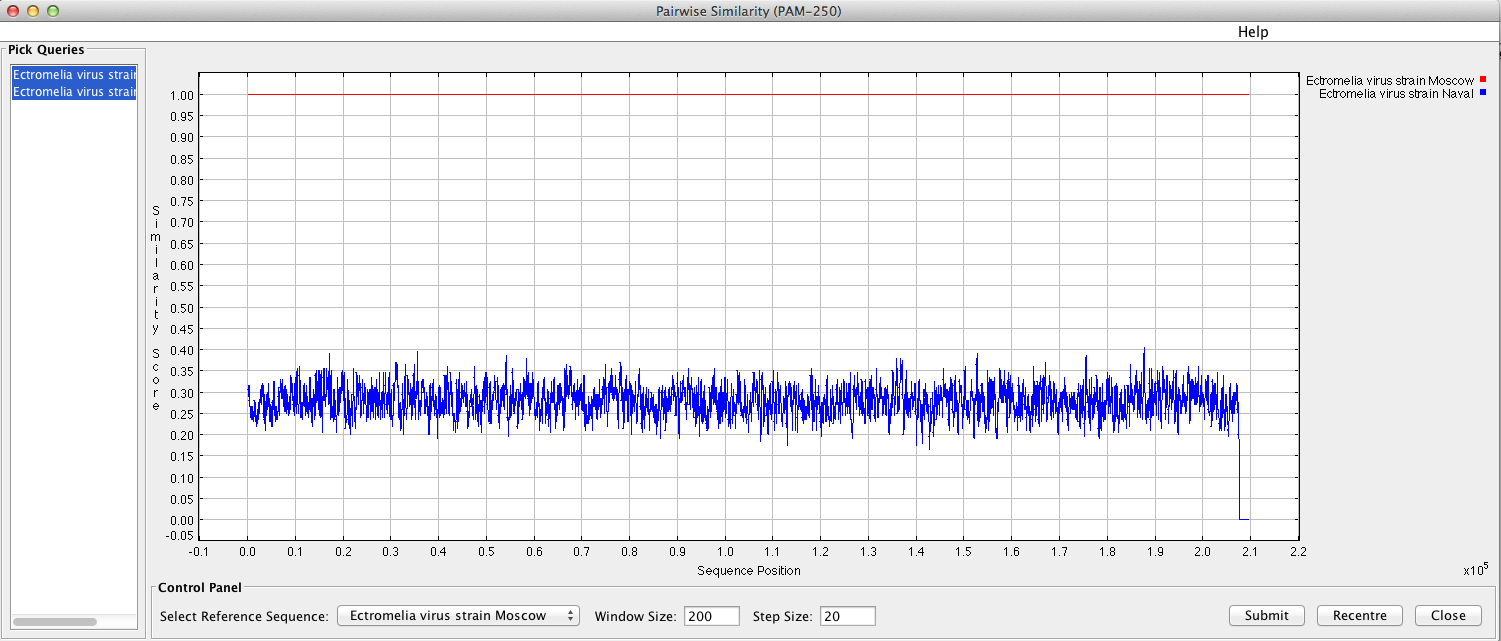
“Sequence Differences Graph” will display a pop-up window with a histogram of all the substitutions for a single sequence. Calculation of differences uses the comparison method selected when the graph was created, and differences are categorized based on the resulting nucleotide. Currently only nucleotide sequences are supported.
“Visual Summary” displays a new window with a detailed view of all insertions, substitutions, deletions, genes, primers and comments for the sequences listed in the sequence window (if “differences” is selected from the drop-down menu on the bottom left of the window). If “nucleotide differences” is selected from the drop-down menu, substitutions will be colored based on the resulting nucleotide (yellow for A, green for C, cyan for G and orange for T), and all other features will be colored black. If “raw conservation” is selected from the drop-down menu, matching residues will be shaded black, similar residues in varying shades of grey and non-conserved residues in white. For protein sequences, it is also possible to select the “scored similarity” views (using either PAM250 or BLOSUM62). If you make changes in the main window that you would like to see reflected in this window, from the “view” menu for this window, select “refresh sequences”. In this window, you may zoom into the image using the “global zoom” slider. It is also possible to export the image and/or change which information is displayed in the window.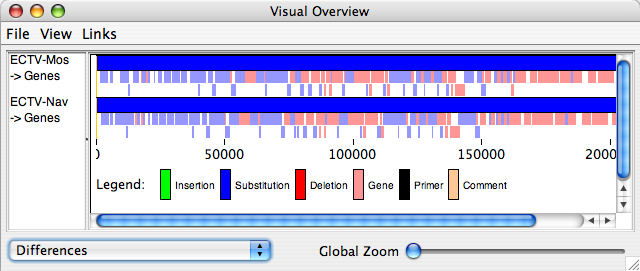
“Percent Identity Table” will display a table of percent identity scores for all sequences against one other according to the current alignment/display of sequences in the sequence window.
“Get Counts” reports the total number of columns in the alignment, as well as the numbers of columns in the alignment that contain: a gap; only 1 nucleotide; 2 different nucleotides, along with the number of columns containing each possible pair of nucleotides; 3 different nucleotides; 4 different nucleotides.
“Get Unique Positions” reports the number of positions in each sequence that are different from ALL other sequences in the alignment.
The Tools Menu
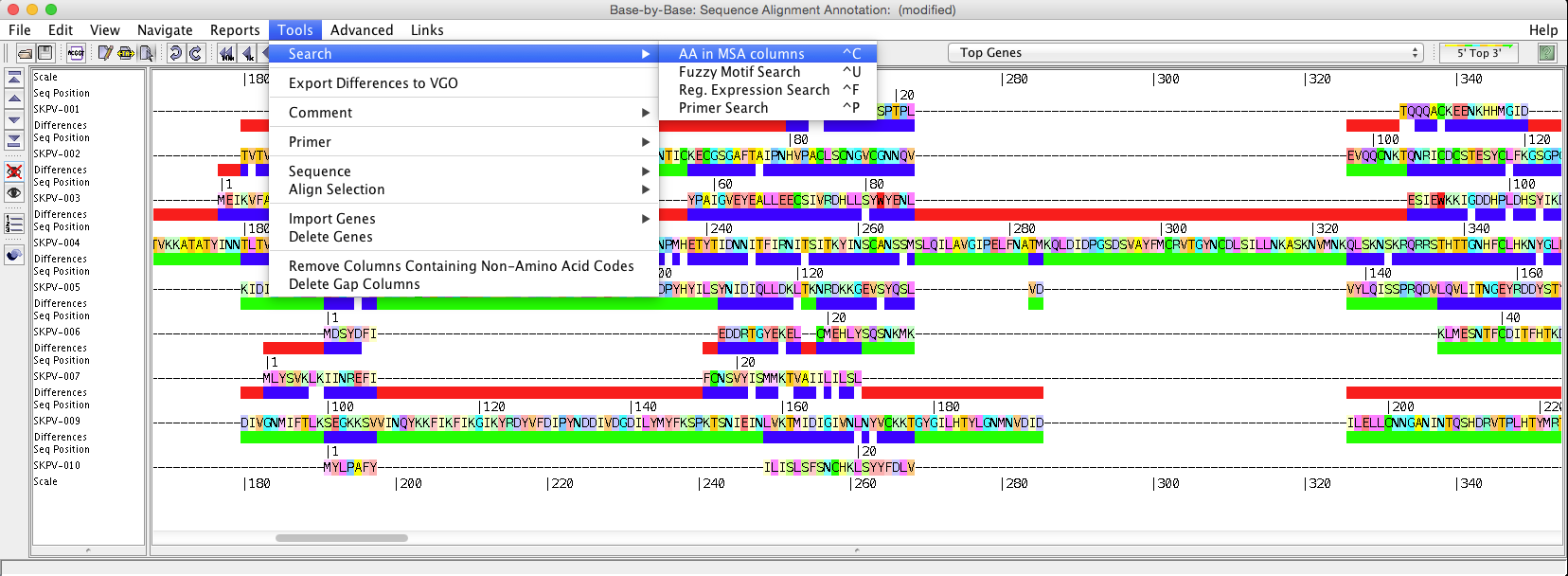
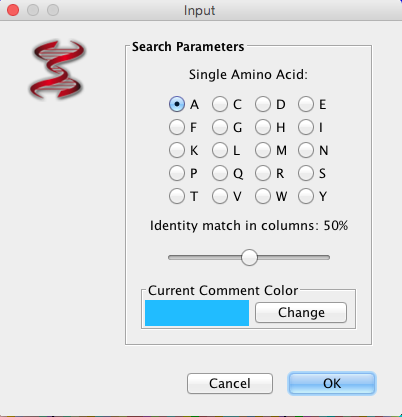
“Search” will allow you to search selected sequences using a “fuzzy motif search”, “regular expression search”, “primer search”, or “AA is MSA (Multiple Sequence Alignment (picture displayed about) column search”. The results of the fuzzy motif or regular expression search will be displayed in a separate window, within which the list can be sorted and also saved. The results displayed are those that correspond to the genome selected on the left-hand side of the results windows. A primer search will conduct a fuzzy motif search within all sequence regions labelled as primers. The search for amino acids in MSA columns is to highlight amino acid positions (within MSA), where conservation level marches % value selected.
“Export Differences to VGO” will export the differences between the sequences you select to a file.
“Comment” allows you to add, remove or edit a comment. You can also search all comments for keywords and list all comments.
“Primer” allows you to add (manually or from a file), remove or edit a primer. You can also search all primers for keywords.
“Sequence” provides simple sequence manipulation and analysis tools:
- “Reverse and Complement” calculates, the reverse, complement, or reverse complement of the selected sequences
- “Calculate A+T%” calculates the A+T% of the selected sequences
- “Calculate ITRs” searches for inverted terminal repeats in the selected sequences
- “Calculate Tandem Repeats” finds tandem repeats in the selected sequences (using etandem)
“Align Selection” aligns the selected portion of the marked sequences in the sequence window using ClustalΩ, Muscle, or Mafft.
Clustal-Omega (Ω): replaces Clustal-W; protein and gene alignement.
Muscle: for fast protein/gene alignment.
Mafft: for alignment of long DNA sequences (eg. >10 kb; poxvirus genome).
“Import Genes” will display a new window from which you can import genes for the genomes in the sequence window either from the VOCs database or from a saved file. If “from VOCs database” is selected, a name search will be conducted to search the VOCs database for matches to each of the sequences. The gene list will then be updated with the imported genes.
“Delete Genes” removes any genes that were added with the “import genes” function.
“The Links Menu” permits you to add or delete links to a list of bookmarks. There is also a link to virology.ca listed here.
“The Help Menu” will link to this online manual for Base-By-Base.
The Advanced Menu
“Pairwise Comparisons” displays a two-dimensional matrix that reports the number of differences between each sequence compared to all others. This feature is useful for finding the number of differences between specific sequences. In the figure below, the differences between sequences 2 and 3 are highlighted in pink, giving 1282 differences, ignoring gaps present in sequence 2. The sequence numbers on the rim of the matrix correspond to the sequence names in the side bar on the left. NOTE: The window states “The differences exclude gaps from the sequence on the right, but not the sequence on the top.” This is incorrect, and should instead state “The differences exclude gaps from the sequence on the right, but not the sequence on the top.”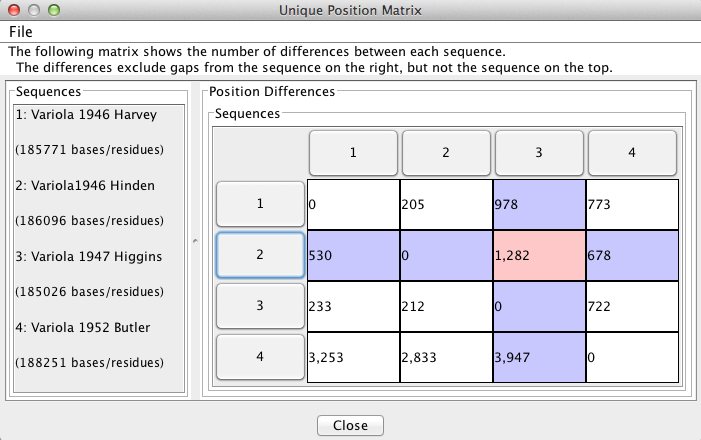
“Find Differences” can be used to compare sequences on a positional basis. The user drags sequences from the “Sequences” box to the “All the same” and “All different” boxes in order to make comparisons. The alignment will be commented at positions where all sequences in the “All the same” column are identical AND all sequences in the “All different”column differ from the first sequence in the “All the same” box.
For example, in the figure below, the alignment will be commented in pink only if 1946 Harvey, 1946 Hinden, and 1952 Butler are identical at the position AND 1947 Higgins is different than 1946 Harvey at the position.Tolerances can be specified for both columns. A log of the the found positions can be viewed and exported to VGO.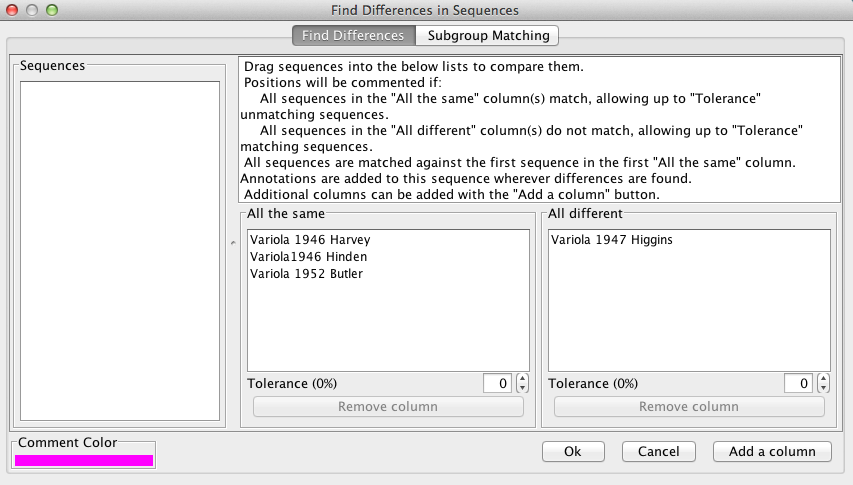
Find Differences Output Help
“Subgroup Matching” can be used to find positions where differences exist between sequences, and works in a similar manner to “Find Differences” (see above). The user drags sequences from the “Sequences” box to the “subgroup” boxes. The aligned sequences will only be commented at positions where all sequences in group 1 differ from each other AND all sequences from group 2 differ from each other. If only one subgroup is desired, the second subgroup box may be left empty. A log of the the found positions can be viewed and exported to VGO.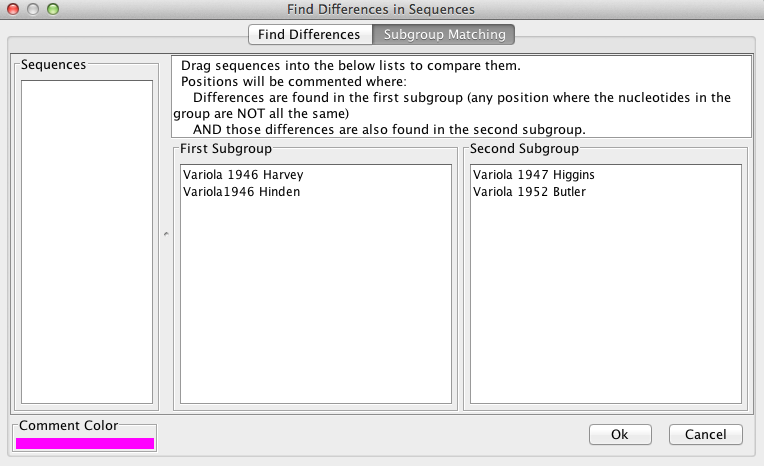
“SNIP” creates a new alignment with SNPs changed to the consensus nucleotide for the given position. SNPs are changed to consensus if there exist more than 2 different nucleotides in a column, or there are fewer SNPs in the column than the specified threshold. The new alignment is opened in an entirely new Base-by-Base window.
“mafft –add” gives access to the mafft command line option “add”. It has the advantage of rapid alignment of additional sequences to a set of already aligned sequences. The original sequences can be imported from a file or aligned normally with BBB.
“View Log“: Once you have run one of the advanced tools (such as Find Differences), the “View Log” option will appear under the “Advanced” menu item. This allows you to view the log for any running or completed process. In the case of Find Differences, you can also plot the results.
Base-By-Base Format
The BBB file format is made based off of the Bioinformatics Sequence Markup Language (BSML).
The BSML Home Page is no longer being maintained, but information and references on the BSML format can still be found at an archived version of the site. Additional information on the BSML specification (including the DTD for the BSML format) can be found here. BSML (and therefore the BBB Format) is a specification built in XML. Base-By-Base files follow a simpler structure than BSML. Below is an example listing containing two sequences with feature data included. The sequence data provided is incomplete to save space.
In order to make things easier, we suggest that users align their files, saving them as Fasta or Clustal files and load them into Base-By-Base. Base-By-Base can then save a BBB format file for later annotation.
Here is an example of annotated sequences:
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Bsml PUBLIC "-//4virology.net//DTD BSML BBB 1.0//EN"
"https://4virology.net...">
<Bsml>
<Definitions>
<Sequences>
<Sequence
comment="Sequence Data Contained in 'Multiple-alignment' section"
id="SEQ-ID:7" length="186103" molecule="dna" title="Variola major virus (Bangladesh-1975)">
<Feature-tables>
<Feature-table title="GENE">
<Feature title="VAR-B-D1L">
<Interval-loc complement="1" endpos="993" startpos="718"/>
<Resource id="GENE-ID:1396"/>
</Feature>
<Feature title="VAR-B-D2L">
<Interval-loc complement="1" endpos="1486" startpos="1055"/>
<Resource id="GENE-ID:1395"/>
</Feature>
<!-- etc -->
</Feature-table>
<Feature-table title="COMMENT">
<Feature comment="fdsafdsa|6666ff|ccffcc" title="COMMENT_TEXT">
<Interval-loc complement="0" endpos="35" startpos="16"/>
</Feature>
</Feature-table>
<Feature-table title="PRIMER">
<Feature class="PRIMER" comment="..." fridge="back"
meltingtemp="18.0" name="..." seq="...">
<Interval-loc complement="0" endpos="9" startpos="1"/>
</Feature>
</Feature-table>
</Feature-tables>
<Sequence
comment="Sequence Data Contained in 'Multiple-alignment' section"
id="SEQ-ID:9" length="185578" molecule="dna" title="Variola virus (India 3 Major, 1967)">
<Feature-tables>
<Feature-table title="GENE">
<Feature title="VAR-I-D1L">
<Interval-loc complement="1" endpos="799" startpos="338"/>
<Resource id="GENE-ID:1751"/>
</Feature>
<Feature title="VAR-I-D2L">
<Interval-loc complement="0" endpos="2232" startpos="1810"/>
<Resource id="GENE-ID:1750"/>
</Feature>
<!-- etc -->
</Feature-table>
</Feature-tables>
</Sequence>
</Sequences>
<Tables>
<Multiple-alignment-table molecule-type="nucleotide">
<Sequence-alignment sequences="2">
<Sequence-data seq-name="Variola major virus (Bangladesh-1975)">TAGTTAGATAAATTAATAATACATAAGTTTTAATACATTAATATTATATT
ATACTATTTTATTTAGTGTCTAGAAAAAAATGTGTGACCCACGACTGTAGGAAACTCTAGAGGGTAAAAAAATCAATCGCTTTATAGAGACCATCAGAAAGAGGTTTAATA
TTTTTGTGAGACCATCGAAGAGAGAAAGAGATAAAACTTTTTACGACTCCATCAGAAAGAGGTTTAATATTTTTGTGAGACCATCGAAGAGAGAAAGAGATAAAACTTTTT
ACGACTCCATCAGAA...</Sequence-data>
<Sequence-data seq-name="Variola virus (India 3 Major, 1967)">----------------------------------------------------
---------------------------------------------------------------------------------------------------------------
---------------------------------------------------------------------------------------------------------------
---------------...</Sequence-data>
<Alignment-consensus/>
</Sequence-alignment>
</Multiple-alignment-table>
</Tables>
<Notebook notebook=""/>
</Definitions>
</Bsml>
Here is an example that shows the same sequences without annotations. Note that these sequences contain the <Feature-tables/> element, which must be removed before the addition of the </Feature-tables> … </Feature-tables> elements or the file will appear corrupted.
<?xml version="1.0" encoding="UTF-8"?>
<!DOCTYPE Bsml PUBLIC "-//4virology.net//DTD BSML BBB 1.0//EN"
"https://4virology.net...">
<Bsml>
<Definitions>
<Sequences>
<Sequence
comment="Sequence Data Contained in 'Multiple-alignment' section"
id="SEQ-ID:7" length="186103" molecule="dna" title="Variola major virus (Bangladesh-1975)">
<Feature-tables/>
<Sequence
comment="Sequence Data Contained in 'Multiple-alignment' section"
id="SEQ-ID:9" length="185578" molecule="dna" title="Variola virus (India 3 Major, 1967)">
<Feature-tables/>
</Sequence>
</Sequences>
<Tables>
<Multiple-alignment-table molecule-type="nucleotide">
<Sequence-alignment sequences="2">
<Sequence-data seq-name="Variola major virus (Bangladesh-1975)">TAGTTAGATAAATTAATAATACATAAGTTTTAATACATTAATATTATATT
ATACTATTTTATTTAGTGTCTAGAAAAAAATGTGTGACCCACGACTGTAGGAAACTCTAGAGGGTAAAAAAATCAATCGCTTTATAGAGACCATCAGAAAGAGGTTTAATA
TTTTTGTGAGACCATCGAAGAGAGAAAGAGATAAAACTTTTTACGACTCCATCAGAAAGAGGTTTAATATTTTTGTGAGACCATCGAAGAGAGAAAGAGATAAAACTTTTT
ACGACTCCATCAGAA...</Sequence-data>
<Sequence-data seq-name="Variola virus (India 3 Major, 1967)">----------------------------------------------------
---------------------------------------------------------------------------------------------------------------
---------------------------------------------------------------------------------------------------------------
---------------...</Sequence-data>
<Alignment-consensus/>
</Sequence-alignment>
</Multiple-alignment-table>
</Tables>
<Notebook notebook=""/>
</Definitions>
</Bsml>