STS Manual
Suffix Tree Searcher (STS) — User Manual
This manual provides a detailed description of the features and settings of STS.
For a brief tutorial on the usage of STS, see the Quick Start Guide.
Table of contents
Introduction
Basic Usage
Input Pane
Output Pane
Introduction
Table of Contents
Suffix Tree Searcher (STS) is a program for performing rapid pattern searches and finding common subsequences in DNA sequence data sets. STS is capable of analyzing small or very large data sets. The Graphical User Interface (GUI) is implemented in Java, and the backend is implemented in C. STS works by indexing the DNA data set into data structures called suffix trees, which reside on the user’s hard disk. The constructed suffix trees can then be explored in a variety of ways. In particular, suffix trees permit extremely fast pattern searches of the data set. The suffix trees can also be analyzed to find the most common patterns (which can be arbitrarily large) occurring in a given data set, based on a variety of filters.
The user interface consists of one window, that is toggled between displaying one of two primary panes: the Input pane and the Output pane. In general, the input pane is used for specify settings and initiate most actions, while the output pane displays the results.
STS accepts FASTA-formatted DNA sequence files as input, and presents output in tabular form.
Basic Usage
Table of Contents
A basic usage scenario works like this: import your sequences, build a suffix tree to index the sequences, analyze the sequences using the suffix tree. There are two distinct types of analyses – searches and traversals.
Searches
Searches consist of querying the data set for exact or inexact patterns. Patterns can be specified by entering a query into the Pattern box of the Input Pane, and clicking the Search button on the Input Pane.
The simplest way to construct a query is to enter a nucleotide sequence (acgt) with optional positional wildcards (*) into the Pattern box, and specify mismatches in the Mismatches box. Multiple queries can be performed at once, by separating the patterns with a semi-colon ( ; ). Searches performed in this manner are conducted on all sequences in the data set, at any position, and any length.
In order search for a pattern located at a specific sequence, and position, enter a triple for each query: <sequence, position, length> e.g. 2, 123500, 15. This will cause the pattern at the given position and sequence to be searched against all sequences.
By default, a search produces a single results table for each query.
Traversals
Traversals consist of examining the input sequences for common substrings. Traversals produce one or more result tables, depending on which settings are selected. The traversal section of the settings window allows the user to select the different types of result tables that are presented. Each different setting will, if selected, produce one or more additional result tables. For example, the user can ask for the longest common substrings (LCS) present N times for N = 2 to N=200, in any number of sequences in the data set, by selecting and setting LCS occurrences=100 in the traversal settings window. In this case, one result table would be produced for each value of N, with the LCS for that N value would displayed in a results table, giving 100 results total for this particular traversal.
Depending on the traversal settings, a single traversal can produce many different result tables – one for each distinct type of traversal that is conducted.
The Input Pane
Table of Contents
The Input pane is where most queries are entered and initiated, and where settings are applied for queries. The Input pane can be divided into four areas: the top bar, the sequence display area, the console output area, and the query input area. The Input pane contains a button to open the settings window.
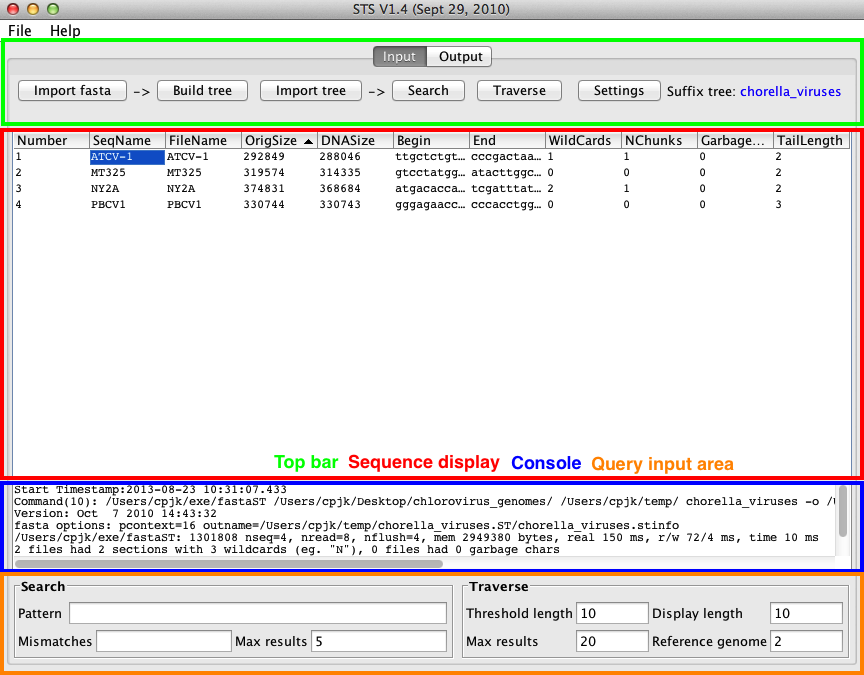
Top bar
The top bar contains buttons for initiating the primary actions of STS.
- Import FASTA – Allows the user to choose one or more FASTA files to import into STS. Sequences must be imported before other actions such as tree building can be taken. Multiple files can be selected using shift-click or cmd-/ctrl-click, and must reside in the same directory. The user is prompted to choose a name for the suffix tree to represent the data set, which by default is the name of the first file selected. This name will also be used for the name of the on-disk file that will contain all suffix tree data for the dataset.
- Build tree – Initiates the construction of the suffix tree index for the input data set. This tree is required in order to query the data set. This process may take several minutes for very large inputs. Settings can be adjusted in the Settings window.
- Import tree – Load a previously constructed suffix tree. Since building the suffix tree is the most time-consuming part of the process, this option allows the user to skip the building phase the next time the program is opened. Consequently, the sequences can be analyzed on different computers without having to rebuild, by transferring the suffix tree data, and importing the tree.
- Search – Initiates a pattern search of the data set. Queries and settings are entered in the search section of the query input area.
- Traverse – Initiates a traversal of the suffix tree. Queries and settings are entered in the traversal section of the query input area. Additional settings are available in the Settings window.
Sequence display area
The sequence display are takes up the most space in the Input panel. It takes the form of a table containing a single row for each sequence in the input files. Each input sequence is associated with a number from 1 to the number of sequences. Information about each sequence is displayed in the following columns, which can be used to sort the sequences:
- Number – The number associated with the sequence.
- SeqName – The name of the sequence as indicated in the FASTA file.
- FileName – The name of the FASTA file that the sequence was imported from.
- OrigSize – The number of bytes read from the file for the sequence.
- DNASize – The number of bytes of the sequence that corresponded to the characters ‘acgt’. All other characters are ignored.
- Begin – The first sixteen nucleotides of the sequence.
- End – The last sixteen nucleotides of the sequence.
- Wildcards – The number of wildcard characters encountered in the sequence. (R, Y, W, S, M, K, H, B, V, D, and N).
- NChunks – The number of contiguous chunks of N characters encountered in the sequence.
- GarbageBytes – The number of non-ACTG non-wildcard characters encountered in the sequence.
- TailLength – The number of repeats of a single motif encountered at the end of the sequence. This column is deprecated; it was previously used for development purposes.
Query input area
The query input area is used to specify the options for searches and traversals before they are initiated through top bar buttons. As such, the area is divided into Search and Traverse areas, each with their own input boxes.
Search
- Pattern – Used to specify search patterns, using ACGT to specify nucleotides, and * to specify wildcards. Patterns can also be specified by entering 3 numbers per pattern for <sequence, position, length> e.g. “2, 125000, 25”. If multiple patterns can be entered by separating individual patterns with a semi-colon “;”.
- Mismatches – This box is used to specify the number of mismatches in the search pattern entered into the Pattern box. If left blank, the number of mismatches allowed will be zero. Mismatches currently do not work when specifying multiple patterns at once.
- Max results – The maximum number of search results to display in non-standard results tables.
Traverse
- Threshold length – Common substrings must be at least this length in order to show up in results tables.
- Display length – Restricts the number of nucleotides that are displayed for each common substring. When copying the sequence from the results table for a pattern search, the entire sequence will still be copied
- Max results – The maximum number of search results to display in non-default result sets.
- Reference genome – Only display results common substrings that are present in this sequence. Takes a sequence number, as shown in the Number column of the Sequence Display Area of the Input Pane.
Console
The console area displays output from the backend executables when building the suffix tree or importing sequences, such as the amount of time a process took to complete, and is used to report any errors that arise during a process. Red text will be shown here if an error occurs.
Settings
The settings window contains tabs for altering the settings of STS:
General settings
Settings pertaining to the general operation of the program.
- Workspace – Specifies the root directory that all files will be written to. This should be the same as the parent directory of the <suffix_tree_name>.ST file, if loading a previously constructed suffix tree.
- Clear results – If selected, STS will delete all results tables in the Output pane every time a new search or traversal is run.
- Advanced – This section is intended primarily for development/debugging purposes. This section allows the user to feed command-line arguments directly to the individual executables that implement the backend.
Build settings
Settings for suffix tree building.
- Partition size (number of nodes) – The number of nodes per suffix tree partition. The average user should not need to adjust this, as the partition size is automatically adjusted for the the user’s computer when the suffix tree is built.
- Suffix buffer size (each) – The buffer size when constructing the suffix tree. The average user should not need to adjust this.
- Skip sort phase – Suffix sorting phase of construction. May increase memory load during the subsequent merge phase. When building suffix trees for large data sets, selecting this option may decrease the time build time.
Traverse settings
Settings governing the types of result sets displayed from a traversal. Each setting will produce additional table(s).
- LCS occurrences – Default set. Gather the longest common substring (LCS) appearing N times, for N=2 to the number specified in any sequence. Produces a single result table.
- LCS inputs – Default set. Gather the single longest substring common K sequences for each value of K from K=2 up to the specified number of inputs. Produces a single result table.
- Number of occurrences – Similar to LCS occurrences, but allows the user to specify the exact values of N, as a comma-separated list, e.g. 2, 3, 5. Produces a result table for each value of N.
- Number of inputs – Similar to LCS inputs, but allows the user to specify the exact values of K, as a comma-separated list, e.g. 2, 3, 4. Produces a result table for each value of N.
- Sets – Allows the user to specify specific subsets of sequences to include a result table for. Each set is specified with curly braces surrounding a comma/space-separated list of the sequences in the set, e.g. {1, 2, 5, 7}. Multiple sets are specified by placing them next to each other, e.g. {1, 2, 5, 7}{1, 4, 6}. A result table will be produced for each set. The number of substrings displayed can be specified by the “Max results” section of the “Traverse” query input section of the Input Panel.
- All singles – Produce a result table for each single sequence.
- All pairs – Produce a result table for each possible pair of sequences.
Search settings
Settings governing the display of search results.
- One result set – If selected, forces results for multi-pattern queries to all be displayed in a single table, rather than separate tables for each pattern.
The Output Pane
Table of Contents
The Output pane is used to view and organize the results tables generated by searches and traversals, and contains a top bar, console, and results display area:
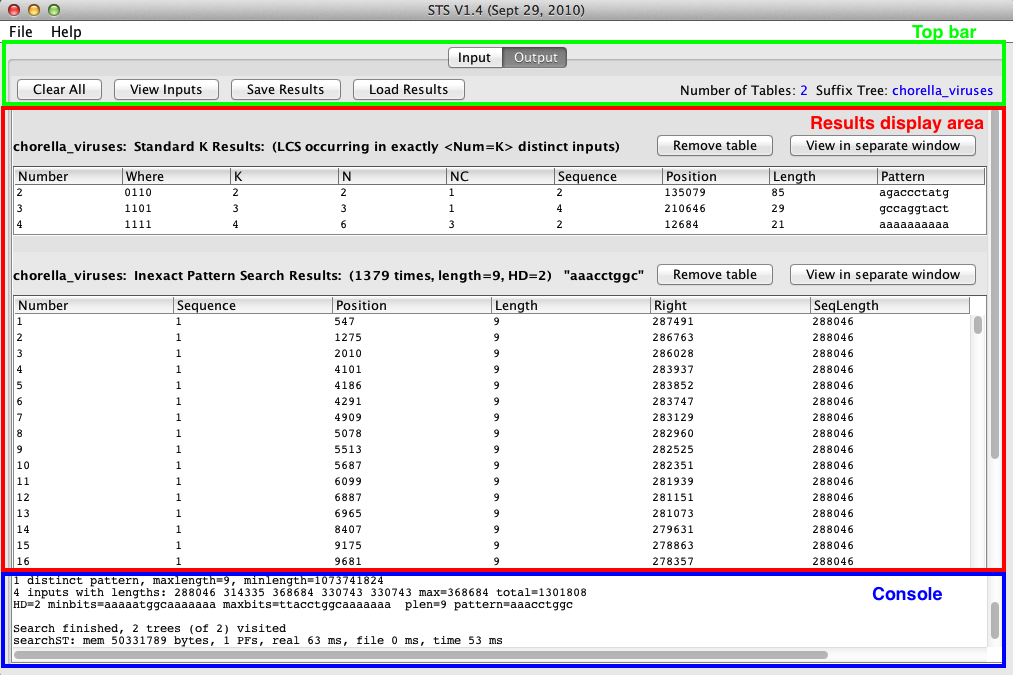
Top bar
- Clear All – Remove all results tables from the display area.
- View Inputs – Open a window that maps sequence numbers to sequence names. This is useful for determining which result corresponds to which sequence(s).
- Save Results – Save the current results tables to disk, so that they can be re-loaded later.
- Load Results – Load previously saved results tables.
Results display area
This area contains result tables from queries. Each row corresponds to a result. Each table has an associated Remove table and View in separate window button. Result tables are added to the display area for each subsequent query, and remain until individually removed, or the window is cleared. Result tables display information about each result in columns. Tables can be sorted by multiple columns. The columns present in each differs depending on whether the table is displaying search or traversal results:
Traversal result tables
Each row in a traversal result table corresponds to a LCS. LCSs are always maximal – they cannot be extended in either direction without violating one or more of their properties. The columns listed below are present in all traversal result tables.
- Number – The hit number. This number serves to identify the specific LCS in the context of the given result table.
- Where – A string of binary digits of length <=32, containing 1’s and 0’s, representing the different input sequences. If there is a 1 in a give position, this indicates that the substring is present in the sequence corresponding to that position, starting at one on the left and counting up for each position to the right. For example, a value of 100110 indicates that a substring is present in sequences 1, 4, and 5.
- K – The number of sequences the substring is present in.
- N – The number of times the substring is present in the number of sequences that the table displays results for.
- NC – The number of other substrings that exist for the given N and length values, since only one is displayed for each length, i.e. In a table that displays only one result for each N-value, if there exist two different sub sequences for N=2, only the longest one will be displayed, but if they are both of the same length, the table will still only display one, and the NC value will be 1 to represent the sequence that is not shown.
- Sequence – One of the input sequences that the substring occurs in.
- Position – The start position of the substring in the sequence specified in the sequence column.
- Length – The length of the substring.
- Pattern – The nucleotide sequence of the substring. The number of nucleotides displayed here is controlled with the “Display length” box in Traverse section of the Input Panel. The user can right-click the pattern to perform a pattern search for the sequence in the whole data set, without the need to copy and paste the pattern into the Input Pane.
Search result tables
Although the number of hits displayed is controlled by the user, the table title always shows the total number of hits.
- Number – The hit number. This number serves to identify the specific result in the context of the given result table.
- Sequence – The sequence that the hit occurred in.
- Position – The position of the hit in the sequence.
- Length – The length of the hit.
- Right – The number of nucleotides remaining to the right (3′) of the hit in given input the sequence.
- SeqLength – The length of the sequence that the hit was found in.
- Count – The total number of hits for the query pattern.
Console
The console area displays output from the backend executables during searches or traversals, such as the amount of time a process took to complete, and is used to report any errors that arise during a process. Red text will be shown here if an error occurs.
Example traversal queries
It is easiest to understand the various traversal result tables in terms of the queries they answer. Below are some example queries, along with the settings required to produce the desired results, and the corresponding result tables that are produced. Note that LCS stands for longest common substring, roughly equivalent to a maximal exact match in other literature. Note also that the letter n refers to a number of times a substring occurs, and the letter k refers to a number of inputs the substring is found in.
-
- Query: What is the single longest common substring occurring exactly n times, for n=2,3,4… 100, without regard to which inputs it may occur in?
- Settings: LCS occurrences = 100.
- Result Table: Standard N Results.
-
- Query: What is the single LCS occurring in exactly k input sequences, for k=2,3,4…8 ?
- Settings: LCS inputs = 8.
- Table: Standard K Results.
-
- Query: What are the S=20 longest (and maximal) substrings found in input 1 and any of the other inputs in a set of 4 sequences?
- Settings: (Reference genome = 1, LCS occurrences = default, LCS inputs = default) OR (sets = {1 2}{1 3}{1 4}, LCS occurrences = default, LCS inputs = default).
- Table: Standard Results OR individual tables for each pair.
-
- Query: What are the S=35 longest (and maximal) substrings occurring exactly n=4 times in the inputs?
- Settings: Number of occurrences = 4. Max results = 35.
- Table: N results: N=4.
-
- Query: What are the S=10 longest (and maximal) substrings occurring at least once in exactly k=5 inputs?
- Settings: Number of inputs = 5, Max results = 10.
- Table: K Results: K=5.
-
- Query: What are the 100 longest common substrings present in input sequence #1 at least twice?
- Settings: Sets = {1}, Max Results=100.
- Table: Custom Results.
Some examples of queries not currently answerable in STS:
- Query: What is the single LCS that present in input 1 for n=1,2,3,4 … ?
- Query: What is the single LCS occurring in at least k inputs, for k=2,3,4… ?
- Query: What is the single LCS occurring in exactly k inputs, for k=1,2,3, in inputs 2,3, and 5? (Variation: Occurring only in {2,3,5}.)
- Query: What are the S=20 longest (and maximal) substrings occurring only in inputs 2 and 3?
- Query: What are the S=20 longest (and maximal) substrings occurring (only) in inputs 2 and 3, but occurring at most (at least?) n=2 times in each?
- Query: What are the S=35 longest (and maximal) substrings occurring at least n=4 times in the inputs?
- Query: What are the S=45 longest (and maximal) substrings occurring in at least k=3 inputs?
Note: There is some inconsistency among the result sets as to whether the numbering commences at 1, or 2. A result row corresponding to 1 usually makes logical sense, but often has no use for the investigator.
Note that Sequence and Position is specific to only one occurrence of the string in question. When there is more than one occurrence (N > 1), it is more-or-less random the value that is chosen for the display.
It can be useful to play with the multi-column sorting feature. For example, sorting on the Where column can often give an investigator a good impression of which inputs are most similar to other inputs. Such information can help guide future search and traversal queries on the same inputs, and may also provide ideas for building new trees with different inputs.
Future Features
Below is a list of some features that we hope to implement in the future. In most cases, the algorithms exist in code or are at least well understood.
- Multiple pattern matching
- Wildcard inexact pattern matching
- Hamming distance inexact pattern matching
- Edit distance inexact pattern matching
- Additional columns of information in result tables
- Table summary information (eg. alphabet of garbage characters, counts, etc.)
- Equivalence, containment, and near-equivalence relations
- Rare and infrequent matches
Other features are on the drawing board, but do not have a clear timeline.
- Selection of input FASTA from multiple sources
- Easy-to-use mix-and-match tree creation
- Reverse, complemented, and reverse-complemented strands
- Better handling of wildcards in indexed sequences
- Client/server architecture
- Integration with the VOCs suite of tools
- Compaction of trees
- Faster handling of repetitive data
There are many other ideas that can be implemented with the help of suffix trees. Gusfield’s book in particular mentions many of these.
- Tandem repeats
- Inexact tandem repeats
- Gene level decomposition of sequences
- Application to larger alphabets (eg. proteins)